
Built to discover
Introducing the first release of CHEMRIYA™ -
a new on-demand chemical space
Meet the Giant

We are proud to present CHEMRIYA™, the next generation chemical space aimed at discovery of new molecules. CHEMRIYA™ has been designed with a simple idea in mind - step by step, make the Chemical Universe tangible and accessible for exploration.
12 Billion Molecules
Vast and Fast
12 billion (and growing!) of novel, synthetically feasible small molecules. Any molecule from CHEMRIYA™ can be ordered and delivered within 4-6 weeks


Navigate Future Drugs
Discovery-ready
An efficient chemical space for drug discovery and development, hit expansion, hit-to lead and lead optimization campaigns. Search for molecule analogs by similarity, pharmacophore, substructure (in progress)
Own your IP Rights
Novel and Unique
IP/patentability - since CHEMRIYA™ molecules are not explicitly listed in the public domain, it’s yours to discover new chemical entities (NCE) and new molecular entities (NME)

Protect your structures
Local and Secure
Strong security - all search queries and results are operated by standalone software on your local computer. No information is visible to us or third parties
Trusted by
Our Clients and Partners









Start Discovering Now
Download chemical space exploration tool to navigate CHEMRIYA™
Stay updated
Unsubscribe at any time. We will keep your email confidential and won't send SPAM.

About
A Dream as Big as Billions of Molecules at Your Fingertips
Imagine billions of uncharted stars in a galaxy waiting to be explored. Imagine billions of feasible molecules holding their unrevealed potential for you to be discovered.
CHEMRIYA™ has been designed with a simple idea in mind - step by step, make the Chemical Universe tangible and easily accessible for exploration.

Created in partnership with BioSolveIT, the first release of the CHEMRIYA™ contains 12 billion accessible on-demand molecules based on 30,000 building blocks and 44 in-house reactions. Several multi-component and ring-closure reactions provide a vast chemical diversity with a broad range of molecular scaffolds.As of 2023, CHEMRIYA™ is composed of 35,000 chemical building blocks and 370 in-house reactionsCHEMRIYA™ is a result of over 25 years of experience and in-house knowledge of synthetic organic and medicinal chemistry, historically accumulated by an expert team of scientists. Operated by an ultra-fast search algorithm, CHEMRIYA™ allows you to investigate untapped areas of tangible molecules.
Highlights:
12 billion (and growing!) of novel, synthetically feasible small molecules. Any molecule from CHEMRIYA™ can be ordered and delivered within 4-6 weeks
An excellent chemical space for drug discovery and development. Perfect for hit expansion, hit-to lead and lead optimization campaigns. Search for molecule analogs by similarity, pharmacophore, substructure (in progress)
IP/patentability - since CHEMRIYA™ molecules are not explicitly listed in the public domain, it’s yours to discover new chemical entities (NCE) and new molecular entities (NME)
Strong security - all search queries and results are operating by standalone software on your local computer. No information is visible to us or third parties
This vast pool of opportunities can be explored with BioSolveIT's Chemical Space navigation platform infiniSee. With utmost precision, infiniSee finds those compounds that are similar to a query molecule, scanning billions of possibilities within seconds. Any structures of interest found in CHEMRIYA™ can be ordered and delivered within 4-6 weeks.We recommend downloading the latest release of infiniSee featuring all the available spaces, including CHEMRIYA™.
Explore CHEMRIYA™ – your imaginary structure could be one click away.

CHEMRIYA Bio™
Explore. Discover. Evaluate.
On top of delivering novel compounds, CHEMRIYA™ allows you to immediately check their activity spectrum in vitro with various bioassays. Any compound you select from CHEMRIYA™ can be synthesized and then tested against your drug target(s) of interest either in fast high-throughput assay or detailed kinetic experiment.
With CHEMRIYA Bio™ services it is possible:
Identify direct, lead small molecule binders/non-binders with Bio-Layer Interferometry
Rank small molecule candidates according to target-based binding parameters
Determine direct, precise binding parameters for select small molecule candidates
Please contact us for more information and a sample quote.

Publications
Comparison of Combinatorial Fragment Spaces and Its Application to Ultralarge Make-on-Demand Compound CatalogsLouis Bellmann, Patrick Penner, Marcus Gastreich, and Matthias Rarey
Journal of Chemical Information and Modeling, 2022, 62 (3), 553-566
https://doi.org/10.1021/acs.jcim.1c01378Abstract

The set of chemical compounds shared by two or more chemical libraries is assessed routinely as means of comparing these libraries for various applications. Traditionally this is achieved by comparing the members of the chemical libraries individually for identity. This approach becomes impractical when operating on chemical libraries exceeding billions or even trillions of compounds in size. As a result, no such analysis exists for ultralarge chemical spaces like the Enamine REAL Space containing over 20 billion compounds. In this work, we present a novel tool called SpaceCompare for the overlap calculation of large, nonenumerable combinatorial fragment spaces. In contrast to existing methods, SpaceCompare utilizes topological fingerprints and the combinatorial character of these chemical spaces. The tool is able to determine the exact overlap of prominent spaces like Enamine’s REAL Space, WuXi’sGalaXi Space, and Otava’s CHEMriya for the first time.
Calculating and Optimizing Physicochemical Property Distributions of Large Combinatorial Fragment SpacesLouis Bellmann, Raphael Klein, and Matthias Rarey
Journal of Chemical Information and Modeling, 2022, 62 (11), 2800-2810
https://doi.org/10.1021/acs.jcim.2c00334Abstract
The distributions of physicochemical property values, like the octanol–water partition coefficient, are routinely calculated to describe and compare virtual chemical libraries. Traditionally, these distributions are derived by processing each member of a library individually and summarizing all values in a distribution. This process becomes impractical when operating on chemical spaces which surpass billions of compounds in size. In this work, we present a novel algorithmic method called SpaceProp for the property distribution calculation of large nonenumerable combinatorial fragment spaces. The novel method follows a combinatorial approach and is able to calculate physicochemical property distributions of prominent spaces like Enamine’s REAL Space, WuXi’sGalaXi Space, and OTAVA’s CHEMriya Space for the first time. Furthermore, we present a first approach of optimizing property distributions directly in combinatorial fragment spaces.
Relevance of the Trillion-Sized Chemical Space “eXplore” as a Source for Drug DiscoveryAlexander Neumann, Lester Marrison, Raphael Klein
ACS Medicinal Chemistry Letters, 2023, 14 (4), 466-472
https://doi.org/10.1021/acsmedchemlett.3c00021Abstract
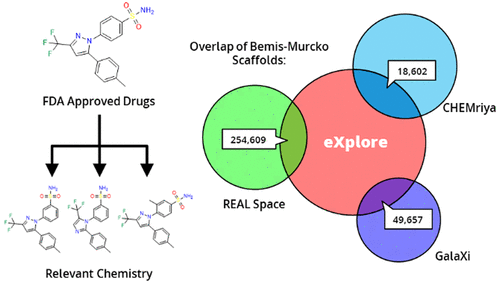
Within the past two decades, virtual combinatorial compound collections, so-called chemical spaces, became an important molecule source for pharmaceutical research all over the world. The emergence of compound vendor chemical spaces with rapidly growing numbers of molecules raises questions about their application suitability and the quality of the content. Here, we examine the composition of the recently published and, so far, biggest chemical space, “eXplore”, which comprises approximately 2.8 trillion virtual product molecules. The utility of eXplore to retrieve interesting chemistry around approved drugs and common Bemis Murcko scaffolds has been assessed with several methods (FTrees, SpaceLight, SpaceMACS). Further, the overlap between several vendor chemical spaces and a physicochemical property distribution analysis has been performed. Despite the straightforward chemical reactions underlying its setup, eXplore is demonstrated to provide relevant and, most importantly, easily accessible molecules for drug discovery campaigns.
Overlap of On-demand Ultra-large Combinatorial Spaces with On-the-shelf Drug-like LibrariesPerebyinis M, Rognan D.
Molecular Informatics, 2023, 42 (1):e2200163
https://doi.org/10.1002/minf.202200163Abstract

On-demand combinatorial spaces are shifting paradigms in early drug discovery, by considerably increasing the searchable chemical space to several billions of compounds while securing their synthetic accessibility. We here systematically compared the on-the-shelf available drug-like chemical space (9 million compounds) to three on-demand ultra-large (ODUL) combinatorial fragment spaces (REAL, CHEMriya, GalaXi) covering 32 billion of readily accessible molecules. Surprisingly, only one space (REAL) intersects almost entirely the currently available drug-like space, suggesting that it is the only ODUL widely suitable for in-stock hit expansion. Of course, expanding a preliminary ODUL hit in the same chemical space is the best possible strategy to rapidly generate structure-activity relationships. All three spaces remain well suited to early hit finding initiatives since they all provide numerous unique scaffolds that are not described by on-the shelf collections.
Maximum Common Substructure Searching in Combinatorial Make-on-Demand Compound SpacesRobert Schmidt, Raphael Klein, Matthias Rarey
Journal of Chemical Information and Modeling, 2022, 62 (9), 2133-2150
https://doi.org/10.1021/acs.jcim.1c00640Abstract

Commercial make-on-demand compound spaces have become increasingly popular within the past few years. Since these libraries are too large for enumeration, they are usually accessed using combinatorial fragment space technologies like FTrees-FS and SpaceLight. Although both search types are of high practical impact, they lack the ability to search for precise structural features on the atomic level. To address this important use case, we developed SpaceMACS enabling efficient and precise maximum common induced substructure (MCIS) similarity and substructure searches within chemical fragment spaces. SpaceMACS enumerates a user-defined number of compounds in a multistep procedure. First, substructures of the query are extracted and matched to all fragments of the space. Then partial results are combined to actual compounds of the space. In this way, SpaceMACS identifies common substructures even if they cross fragment borders. We applied SpaceMACS on three commercial fragment spaces searching for the 150 000 most similar analogs to a glucosyltransferase binder from literature. We were able to find almost all building blocks used for the synthesis of the 90 listed analogs and a plethora of additional results. SpaceMACS is the missing link to enable rational drug discovery on make-on-demand combinatorial catalogs. No matter whether initial compound suggestions come from a de novo design, an AI-based compound generation, or a medicinal chemist’s drawing board, the method gives access to the structurally closest chemically available analogs in seconds to at most minutes.
Computational approaches streamlining drug discoverySadybekov, A.V., Katritch, V.
Nature, 2023, 616, 673–685
https://doi.org/10.1038/s41586-023-05905-zAbstract
Computer-aided drug discovery has been around for decades, although the past few years have seen a tectonic shift towards embracing computational technologies in both academia and pharma. This shift is largely defined by the flood of data on ligand properties and binding to therapeutic targets and their 3D structures, abundant computing capacities and the advent of on-demand virtual libraries of drug-like small molecules in their billions. Taking full advantage of these resources requires fast computational methods for effective ligand screening. This includes structure-based virtual screening of gigascale chemical spaces, further facilitated by fast iterative screening approaches. Highly synergistic are developments in deep learning predictions of ligand properties and target activities in lieu of receptor structure. Here we review recent advances in ligand discovery technologies, their potential for reshaping the whole process of drug discovery and development, as well as the challenges they encounter. We also discuss how the rapid identification of highly diverse, potent, target-selective and drug-like ligands to protein targets can democratize the drug discovery process, presenting new opportunities for the cost-effective development of safer and more effective small-molecule treatments.
Recent Developments in Structure-Based Virtual Screening ApproachesChristoph Gorgulla
arXiv:2211.03208v1
https://arxiv.org/abs/2211.03208
https://doi.org/10.48550/arXiv.2211.03208Abstract
Drug development is a wide scientific field that faces many challenges these days. Among them are extremely high development costs, long development times, as well as a low number of new drugs that are approved each year. To solve these problems, new and innovate technologies are needed that make the drug discovery process of small molecules more timea and cost-efficient, and which allow to target previously undruggable target classes such as protein-protein interactions. Structure-based virtual screenings have become a leading contender in this context. In this review, we give an introduction to the foundations of structure-based virtual screenings, and survey their progress in the past few years. We outline key principles, recent success stories, new methods, available software, and promising future research directions. Virtual screenings have an enormous potential for the development of new small-molecule drugs, and are already starting to transform early-stage drug discovery.
Exploration of Ultralarge Compound Collections for Drug DiscoveryWendy A. Warr, Marc C. Nicklaus, Christos A. Nicolaou, and Matthias Rarey
Journal of Chemical Information and Modeling, 2022, 62 (9), 2021-2034
https://doi.org/10.1021/acs.jcim.2c00224Abstract

Designing new medicines more cheaply and quickly is tightly linked to the quest of exploring chemical space more widely and efficiently. Chemical space is monumentally large, but recent advances in computer software and hardware have enabled researchers to navigate virtual chemical spaces containing billions of chemical structures. This review specifically concerns collections of many millions or even billions of enumerated chemical structures as well as even larger chemical spaces that are not fully enumerated. We present examples of chemical libraries and spaces and the means used to construct them, and we discuss new technologies for searching huge libraries and for searching combinatorially in chemical space. We also cover space navigation techniques and consider new approaches to de novo drug design and the impact of the “autonomous laboratory” on synthesis of designed compounds. Finally, we summarize some other challenges and opportunities for the future.
Navigating large chemical spaces in early-phase drug discoveryMalte Korn, Christiane Ehrt, Fiorella Ruggiu, Marcus Gastreich, Matthias Rarey
Current Opinion in Structural Biology, 2023, 80:102578
https://doi.org/10.1016/j.sbi.2023.102578Abstract
The size of actionable chemical spaces is surging, owing to a variety of novel techniques, both computational and experimental. As a consequence, novel molecular matter is now at our fingertips that cannot and should not be neglected in early-phase drug discovery. Huge, combinatorial, make-on-demand chemical spaces with high probability of synthetic success rise exponentially in content, generative machine learning models go hand in hand with synthesis prediction, and DNA-encoded libraries offer new ways of hit structure discovery. These technologies enable to search for new chemical matter in a much broader and deeper manner with less effort and fewer financial resources. These transformational developments require new cheminformatics approaches to make huge chemical spaces searchable and analyzable with low resources, and with as little energy consumption as possible. Substantial progress has been made in the past years with respect to computation as well as organic synthesis. First examples of bioactive compounds resulting from the successful use of these novel technologies demonstrate their power to contribute to tomorrow's drug discovery programs. This article gives a compact overview of the state-of-the-art.

Download
Download software to navigate CHEMRIYA™
Entire chemical space of CHEMRIYA™ is available to navigate through infiniSee software from BioSoveIT. infiniSee enables similarity searching, based on a query molecule, through billions of compounds in chemical spaces.

Contact Us
Contact information:
5000 Yonge Street, Suite 1901
Toronto, Ontario M2N 7E9
CanadaTel: +1-855-745-5570
Fax: +1-866-608-0550Email: info@chemriya.com
Website: https://chemriya.com
Please send us a message:
Thank you!
Your message has been sent. We'll be in touch shortly.
Malesuada
Integer eget aliquet nibh praesent tristique magna. Sit amet justo donec enim.
Egestas congue
Commodo ullamcorper a lacus vestibulum sed arcu non odio. Urna et pharetra pharetre.
Feugiat pretium
Quis eleifend quam adipiscing vitae proin sagittis nisl rhoncus diam placerat mattis.